DinuQ
The DinuQ (Dinucleotide Quantification) Python3 package provides a range of metrics for quantifying nucleotide, dinucleotide and synonymous codon representation in genetic sequences. These include the recently developed corrected Synonymous Dinucleotide Usage (SDUc) and corrected Relative Synonymous Dinucleotide Usage (RSDUc).
In this documentation page you can find out how to use the DinuQ package for analysing the dinucleotide content in your genetic sequences as well as read about the maths behind the metrics implemented in the package.
Installation
Using pip, in a Unix terminal do: pip install dinuq
Then in python do: import dinuq
RDA
Relative dinucleotide abundance - dinuq.RDA()
RDA Maths
RDA is the traditional metric for quantifying dinucleotide representation. It simply represents the ratio between the frequency of a given dinucleotide (e.g. frequency of CpG) over the product of its components’ individual frequencies (frequency of Cs times frequency of Gs) in a nucleotide sequence.
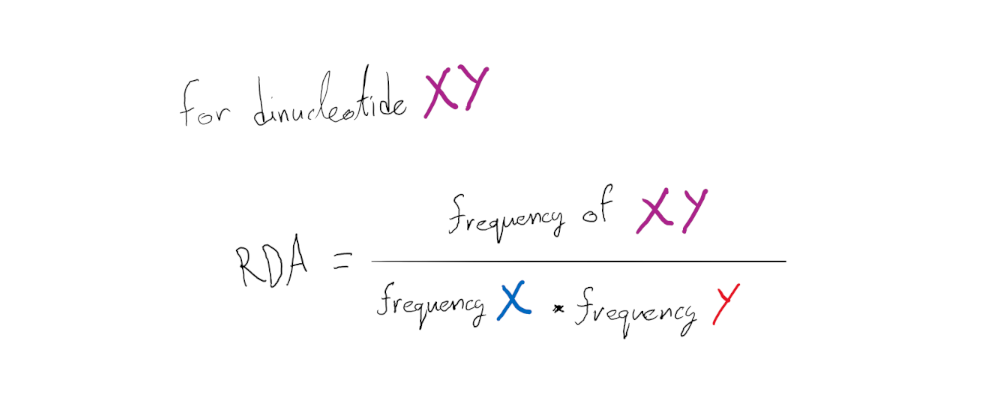
If the dinucleotide frequency is equal to that expected from its components’ individual frequencies then the RDA should have a value of 1 (no bias in the dinucleotide representation). Similarly, RDA values below 1 suggest under-representation of the given dinucleotide in the sequence and values over 1 over-representation.
RDA Usage
rda = dinuq.RDA( fasta_file, dinucl, position )
Required arguments:
-
fasta_file: a fasta file containing any number of sequences with different headers. -
dinucl: A list of dinucleotides of interest (still needs to be a list if it’s only one, e.g. [‘CpG’]). If you’d like to calculate the metric for all dinucleotide combinations use the premade listdinuq.alldnsinstead of writing everything youself (see Useful dictionaries/lists below). Note that Us are used instead of Ts as a convention (e.g. UpA instead of TpA).
Optional arguments:
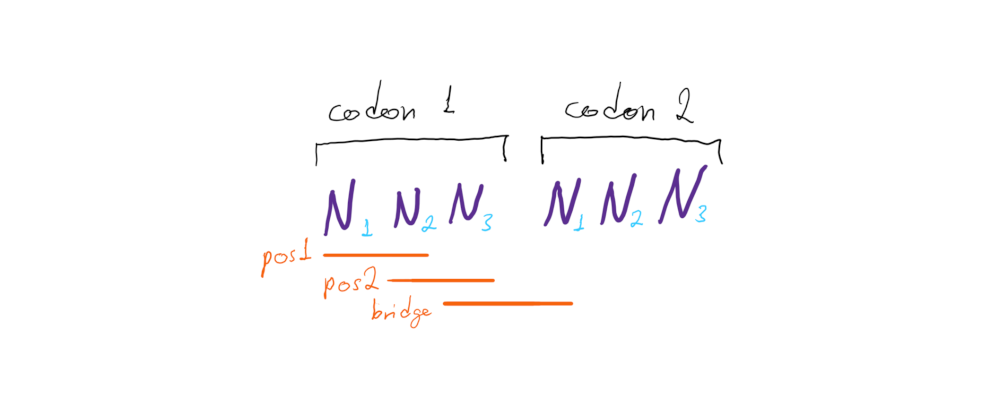
position: By default, the module will calculate a single RDA value for each sequence (position = [‘all’]). If you have a coding sequence, you could choose to calculate a separate RDA value for each coding frame dinucleotide position; for example in the coding sequence ATG CGG dinucleotides AT and CG are found in frame position 1, TG and GG in position 2 and GC in the bridge position between the codons. To use this option simply provide a list of the positions you want to calculate RDA for asposition = ['pos1', 'pos2', 'bridge'].
Output
The output of the module is a dictionary of headers as keys and contained dictionaries as values. The contained dictionaries each have dinucleotides (or dinucleotides with frame positions e.g. CpGbridge) as keys and the corresponding RDA value as the value.
rda = { 'accession': { 'dinucleotideposition': rda_value } }
Exporting output
You can easily export the dictionary output into a tabular file using
dinuq.RDA_to_tsv(). The module’s required arguments are:
- A rda dictionary produced by the RDA module.
- A name for the output file.
RDA_to_tsv( rda_dic, output_name )
The output will be tab-delimited (tsv) by default, but you can change the
delimiter with the optional argument sep, e.g.
RDA_to_tsv( rda_dic, output_name, sep=',' )
Example Usage
>>>import dinuq as dn
>>>rda = dn.RDA( 'myseqs.fas', [ 'CpG', 'UpA', 'CpC' ], [ 'pos1', 'pos2', 'bridge' ] )
>>>dn.RDA_to_tsv( rda, 'myseqs_RDA.tsv' )
SDUc
corrected Synonymous Dinucleotide Usage - dinuq.SDUc()
SDUc Maths
The SDUc is a novel metric for quantifying dinucleotide representation, accounting for the codon and amino acid usage in a given coding sequence. SDUc can only be calculated for coding sequences, but has certain advantages compared to RDA including estimating a distribution around the null expectation of each dinucleotide’s representation that can be used for statistically assessing the under- or over-representation of the dinucleotide of interest.
The SDUc calculation
Step 1: calculate the expected codon frequency
The SDUc framework is based on comparing the dinucleotide usage of synonymous codons in a coding sequence to that expected when the sequence’s codon usage reflects its single nucleotide composition (null expectation).
To first assess the null expectation, we calculate the frequency of each codon expected by the sequence’s nucleotide composition:
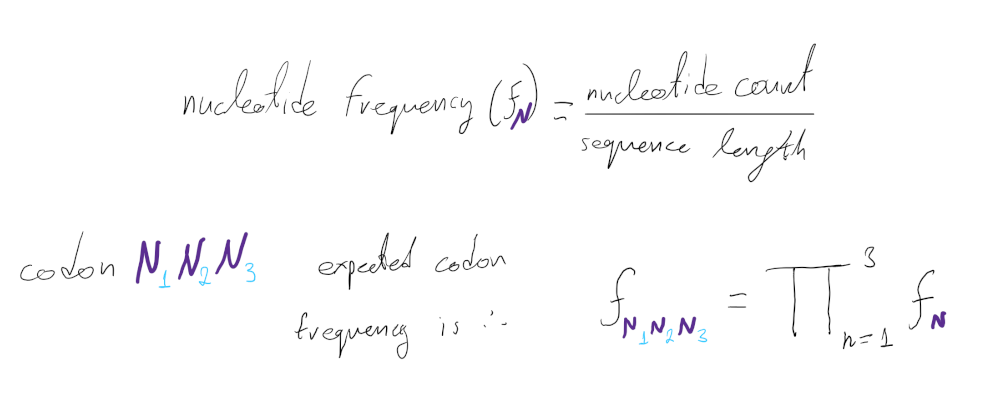
Since only 61 out of the 64 triplet combinations are coding, we need to correct the frequency calculations to exclude the 3 stop codons:

Now that we have calculated the set of all corrected expected codon frequencies (c’) we can use them to derive a null dinucleotide expectation for any dinucleotide in any frame position.
Step 2: Calculate SDUc values
Instead of calculating a single value for each dinucleotide (like with the RDA), a different SDUc value needs to be calculated for each frame dinucleotide position in the coding sequence: position 1 (first and second nucleotides of the codon), positions 2 (second and third codon nucleotides), and bridge (the third nucleotide of the first codon and first nucleotide of the second codon):
Then the SDUc value is defined as follows for:
- a dinucleotide j
- a dinucleotide frame position h
- an amino acid (or amino acid pair in the case of the bridge position) i
- the number of occurrences of an amino acid (or pair) ni
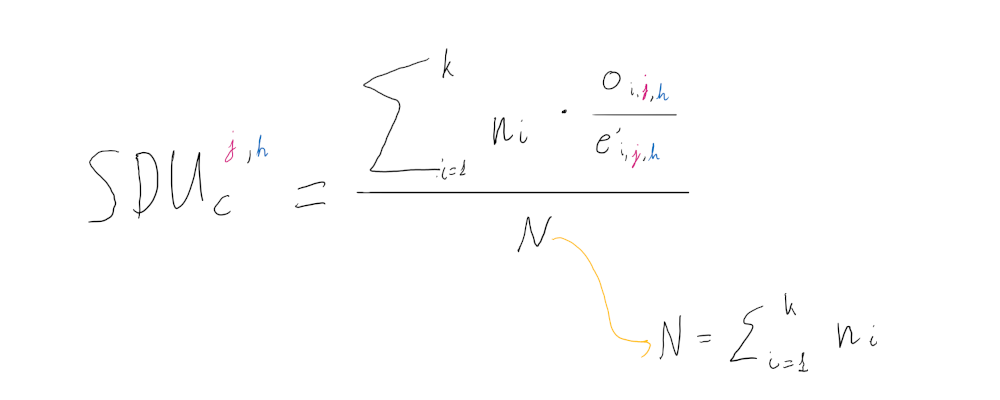
with k being the set of informative amino acids (or pairs) in the sequence translation.
Thus the SDUc metric is essentially a weighted average of the ratio between the two following parameters:
-
oi,j,h: the synonymous proportion of dinucleotide j in frame position h observed in the sequence
-
e’i,j,h: the expected synonymous frequency of dinucleotide j in frame position h given the set of corrected expected codon frequencies c’
for all informative amino acids (or pairs) in set k.
What defines if an amino acid is informative for a given dinucleotide/frame position combination is whether it can be encoded by synonymous codons with more than one dinucleotide combinations in the given frame position. For example, arginine (R) can be encoded by 6 different codons, four of which have a CpG and 2 that have a ApG in frame position 1. This means that arginine is an informative amino acid for CpGpos1 and ApGpos1. On the contrary, proline (P) can be encoded by four codons that all have CpC in position 1, so proline is not informative for frame position 1 with any dinucleotide. Due to this limitation no SDUc value can be calculated for the following dinucleotide/position combinations: CpCpos1, CpApos1, GpCpos1, GpGpos1, GpUpos1, GpApos1, UpGpos1, UpApos1, ApCpos1, ApUpos1, ApApos1.
Step 3: Estimate the null distribution
A valuable feature of the SDUc framework is that, since the amino acid composition of the sequence is taken into account, one can estimate the error around the metric’s null distribution (a value of 1 in the case of the SDUc) for any given sequence. This is done by simulating a (user-specified) number of sequences that produce the exact same protein sequence as the starting (real) coding sequence, but have codons sampled based on the corrected expected frequencies calculated using the single nucleotide composition of the starting sequence (set c’). For example:
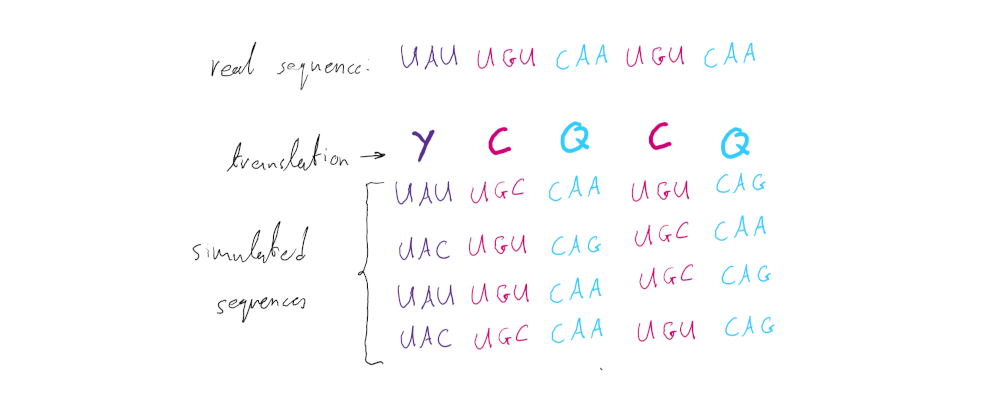
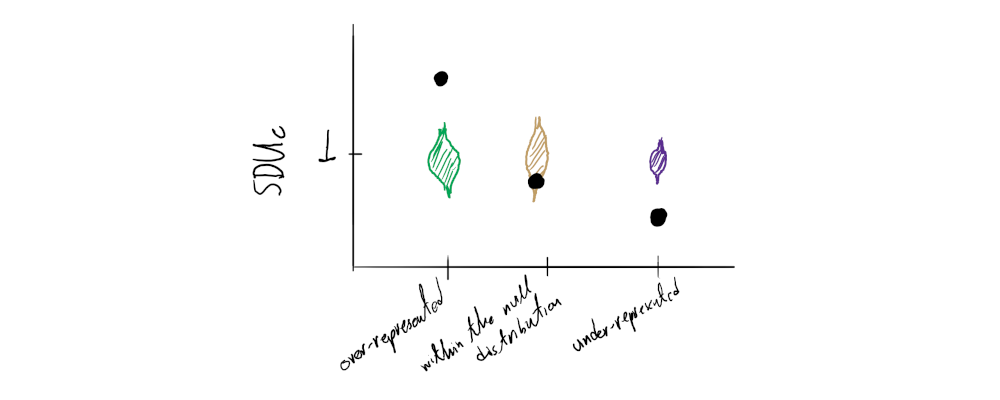
These mock sequences should all theoretically represent the null expectation of the metric (no dinucleotide bias) and - if they were to be of infinite length - should always give a SDUc value of 1. By calculating the SDUc value for every simulated sequence we can create a normal distribution around the mean of 1. Thus, instead of simply comparing the real sequence’s SDUc value to a single number (1) we are able to statistically assess whether it falls within the null distribution’s confidence intervals or not.
SDUc Usage
sduc = dinuq.SDUc( fasta_file, dinucl, position, boots, custom_nt )
Required arguments:
-
fasta_file: a fasta file containing any number of coding sequences with different headers. -
dinucl: A list of dinucleotides of interest (still needs to be a list if it’s only one, e.g. [‘CpG’]). If you’d like to calculate the metric for all dinucleotide combinations use the premade listdinuq.alldnsinstead of writing everything youself (see Useful dictionaries/lists below). Note that Us are used instead of Ts as a convention (e.g. UpA instead of TpA).
Optional arguments:
position: By default the module will calculate SDUc values for all three frame positions (pos1, pos2, bridge). If you want to calculate the metric for specific positions simply provide a list of the desired frame positions, e.g.position = ['bridge'].boots: If you want to calculate confidence intervals around the null expectation, you can specify a number of iterations for the sequence simulation method described above (suggested values are 100 for long coding sequences and up to 1000 for shorter sequences). Notice that this will significantly slow down the calculation.custom_nt: By default the single nucleotide composition of the given coding sequence will be used for esimating the expected codon frequencies, however you can also specify a custom single nucleotide compositions profile to base the expected dinucleotide usage on for each accession in your fasta file (e.g. if you want to calculate SDUc values for all genes in a genome, given the nt composition of the entire genome, instead of the nt composition of each gene). This should be provided as a dictionary formatted as shown here:{ 'acc1': { 'G': fG, 'C': fC, 'A': fA, 'T': fT }, 'acc2': ... }
Output
The output of the module is a dictionary of headers as keys and contained dictionaries as values. The contained dictionaries have each dinucleotide position as keys (e.g. CpGbridge) and a list of calculated SDUc values as the value. If a null distribution is being calculated, a contained list of SDUc values calculated for each simulated sequence is included.
sduc = { 'accession': { 'dinucleotideposition': [ sdu_value, [ bootstrap_value1, bootstrap_value2, bootstrap_valuen ] ] } }
Exporting output
You can easily export the dictionary output into a tabular file using
dinuq.dict_to_tsv(). The module’s required arguments are:
- A sduc dictionary produced by the SDUc module.
- A name for the output file.
The output will be tab-delimited (tsv) by default, but you can change the
delimiter with the optional argument sep, e.g.
dict_to_tsv( dictionary, output_name, sep=',' )
If the boots option has been used in the SDUc module to calculate the null distribution,
the dict_to_tsv() module will automatically include columns with the 95% confidence intervals
of the distribution for each SDUc value. The single standard deviation intervals and minimum/maximum
values of the distribution can also be produced with the optional argument error, e.g.
dict_to_tsv( dictionary, output_name, error='stdev' )
or
dict_to_tsv( dictionary, output_name, error='extrema' )
Example Usage
>>>import dinuq as dn
>>>sduc = dn.SDUc( 'myseqs.fas', [ 'CpG', 'UpA', 'CpC' ], [ 'pos1', 'pos2', 'bridge' ], boots=100 )
>>>dn.dict_to_tsv( sduc, 'myseqs_SDUc.tsv' )
RSDUc
corrected Relative Synonymous Dinucleotide Usage - dinuq.RSDUc()
RSDUc Maths
The RSDUc is an extension of the SDUc metric, where all values (for any sequence and dinucleotide/position combination) are directly comparable to one another.
Because of the way SDUc is calculated the null expectation for all dinucleotide/position combinations is always SDUc = 1 ( oi,j,h = e’i,j,h ). However different dinucleotide/position combinations have different maximum SDUc values. This means that, theoretically, the SDUc values of different sequences or dinucleotide/position combinations are not directly comparable to one another, e.g. a CpGpos1 SDUc value being lower than a UpApos2 SDUc value does not necessarily mean that the relative representation of CpGs in frame position 1 is less than the relative representation of UpAs in position 2 of the sequence.
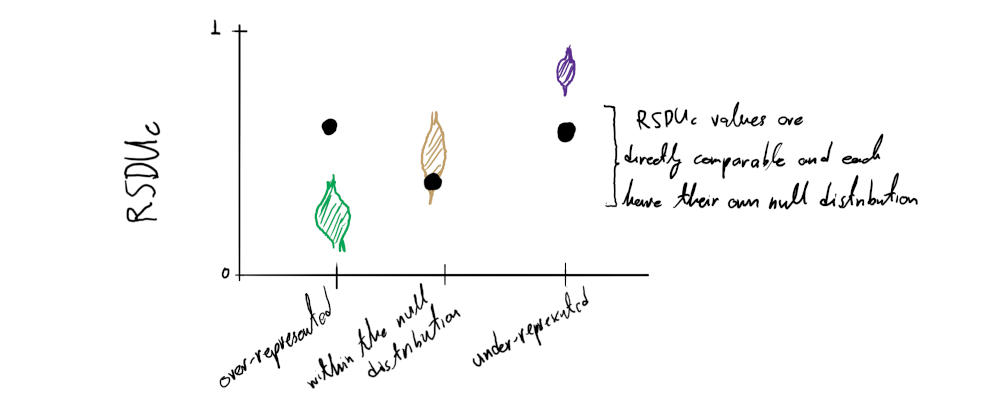
The RSDUc accounts for this problem, by simply dividing the SDUc value by the maximum SDUc value that the given sequence can take for the dinucleotide/position combination of interest, where oi,j,h = 1:
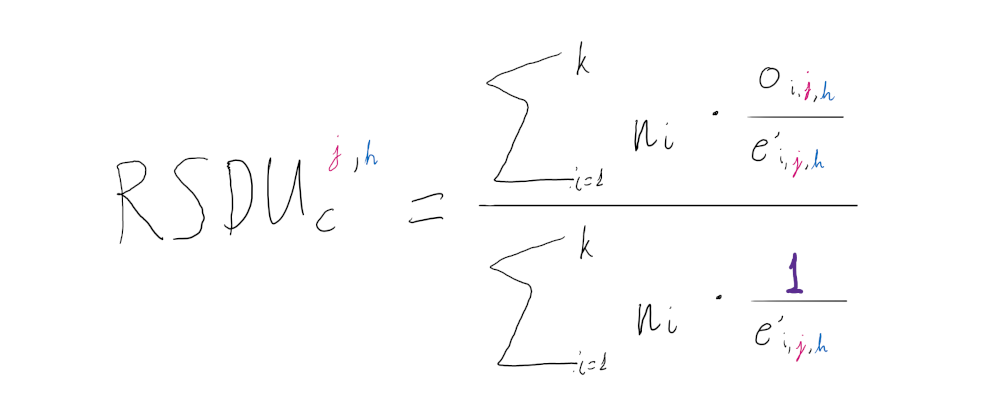
In this way all RSDUc values of any dinucleotide/position combination calculated for any sequence are directly comparable and bound by 0 and 1, but now they do not have the same null expectation. Using the same method for estimating the null distribution for each SDUc value described above (Step 3: Estimate the null distribution) we can determine that null expectation. So RSDUc can be used for both assessing if a dinucleotide is over- or under-represented in a sequence and compare the relative representation to other sequences, frame positions or dinucleotides:
RSDUc Usage
rsduc = dinuq.RSDUc( fasta_file, dinucl, position, boots, custom_nt )
Required arguments:
-
fasta_file: a fasta file containing any number of coding sequences with different headers. -
dinucl: A list of dinucleotides of interest (still needs to be a list if it’s only one, e.g. [‘CpG’]). If you’d like to calculate the metric for all dinucleotide combinations use the premade listdinuq.alldnsinstead of writing everything youself (see Useful dictionaries/lists below). Note that Us are used instead of Ts as a convention (e.g. UpA instead of TpA).
Optional arguments:
position: By default the module will calculate RSDUc values for all three frame positions (pos1, pos2, bridge). If you want to calculate the metric for specific positions simply provide a list of the desired frame positions, e.g.position = ['bridge'].boots: If you want to calculate confidence intervals around the null expectation, you can specify a number of iterations for sequence simulation method described above (suggested values are 100 for long coding sequences and up to 1000 for shorter sequences). Notice that this will significantly slow down the calculation.custom_nt: By default the single nucleotide composition of the given coding sequence will be used for esimating the expected codon frequencies, however you can also specify a custom single nucleotide compositions profile to base the expected dinucleotide usage on for each accession in your fasta file (e.g. if you want to calculate SDUc values for all genes in a genome, given the nt composition of the entire genome, instead of the nt composition of each gene). This should be provided as a dictionary formatted as shown here:{ 'acc1': { 'G': fG, 'C': fC, 'A': fA, 'T': fT }, 'acc2': ... }
Output
The output of the module is a dictionary of headers as keys and contained dictionaries as values. The contained dictionaries have each dinucleotide position as keys (e.g. CpGbridge) and a list of calculated SDUc values as the value. If a null distribution is being calculated, a contained list of SDUc values calculated for each simulated sequence is included.
rsduc = { 'accession': { 'dinucleotideposition': [ rsdu_value, [ bootstrap_value1, bootstrap_value2, bootstrap_valuen ] ] } }
Exporting output
You can easily export the dictionary output into a tabular file using
dinuq.dict_to_tsv(). The module’s required arguments are:
- A rsduc dictionary produced by the RSDUc module.
- A name for the output file.
The output will be tab-delimited (tsv) by default, but you can change the
delimiter with the optional argument sep, e.g.
dict_to_tsv( dictionary, output_name, sep=',' )
If the boots option has been used in the RSDUc module to calculate the null distribution,
the dict_to_tsv() module will automatically include columns with the 95% confidence intervals
of the distribution for each RSDUc value. The single standard deviation intervals and minimum/maximum
values of the distribution can also be produced with the optional argument error, e.g.
dict_to_tsv( dictionary, output_name, error='stdev' )
or
dict_to_tsv( dictionary, output_name, error='extrema' )
Example Usage
>>>import dinuq as dn
>>>rsduc = dn.RSDUc( 'myseqs.fas', [ 'CpG', 'UpA', 'CpC' ], [ 'pos1', 'pos2', 'bridge' ], boots=100 )
>>>dn.dict_to_tsv( rsduc, 'myseqs_RSDUc.tsv' )
RSCU
dinuq.RSCU()
The RSCU module will calculate the Relative Synonymous Codon Usage for all sequences in a given fasta file.
Required arguments:
fasta_file: a fasta file containing any number of coding sequences with different headers.
Output
The output of the module is a dictionary of accessions as keys and contained dictionaries as values. The contained dictionaries have each codon as keys and the calculated RSCU value as the value.
rscu = { 'accession': { 'codon': rscu_value } }
Exporting output
You can export the dictionary output into a tabular file using
dinuq.RSCU_to_tsv(). The module’s required arguments are:
- A rscu dictionary produced by the RSCU module.
- A name for the output file.
The output will be tab-delimited (tsv) by default, but you can change the
delimiter with the optional argument sep, e.g.
dinuq.RSCU_to_tsv( rscu, output_name, sep=',' )
Accessory modules
cod_check
This module checks if there are any sequences in a fasta file that:
- are non-coding
- have internal stop codons
- have ambiguous nucleotides
The module will print out the problems to standard output and return a list of headers in the fasta file that will cause problems when calculating metrics that require coding sequences.
Example Usage
prob_seqs = dinuq.cod_check( fasta_file )
To include sequences with ambiguous nucleotides (that are coding) in the list,
use the optional argument errors = 'all'.
ntcont
This module calculates the single nucleotide composition of all sequences in a fasta file.
Output is given as a dictionary as follows:
{ acc1:{ 'G':Gcon, 'C':Ccon, 'A':Acon, 'T':Tcon }, acc2:... }
Example Usage
nt_content = dinuq.ntcont( fasta_file )
eprimeall
For advanced users, this module can make the following internal calculations used for the final SDUc calculation:
- dictionary of corrected expected dinucleotide frequencies for any set of dinucleotide/position combinations (extras = ‘eprime’ - default)
- dictionary of corrected expected codon frequencies (extras = ‘codonfreq’)
- stop codon correction factor (extras = ‘correction’)
The required input can be either a fasta file, or a dictionary of single nucleotide
compositions, formatted like the output of dinuq.ntcont().
Example Usage
>>>import dinuq as dn
>>>eprime_values = dn.eprimeall( 'myseqs.fas', ['CpG', 'UpA', 'CpC'], ['pos1', 'pos2', 'bridge'] )
>>>codon_freqs = dn.eprimeall( nt_content, [], [], extras = 'codonfreq' )
>>>correction_f = dn.eprimeall( 'myseqs.fas', [], [], extras = 'correction' )
Useful dictionaries/lists
-
dinuq.sycoDictionary of amino acid monograms as keys and lists of respective codons as values. -
dinuq.cod_aa_dicDictionary of codons as keys and respective amino acid monograms as values. -
dinuq.alldnsList of all dinucleotides in the format compatible with DinuQ (e.g. UpA).
Citations
Please cite the following work when using DinuQ:
- Lytras, S.; Hughes, J. (2020) Synonymous Dinucleotide Usage: A Codon-Aware Metric for Quantifying Dinucleotide Representation in Viruses. Viruses, 12:462.
- MacLean, O.#; Lytras, S.#; Weaver, S.; Singer, J.B.; Boni, M. F.; Lemey, P.; Kosakovsky Pond, S.L.; Robertson, D.L. (2021) Natural selection in the evolution of SARS-CoV-2 in bats created a generalist virus and highly capable human pathogen. PLoS Biol, 19(3):e3001115.